




Evaluar las fuentes de información empleadas por los Registros Autonómicos de Enfermedades Raras (RAER) para la captación de la enfermedad de Wilson en España, calcular su prevalencia y mortalidad, y describir las características sociodemográficas de las personas afectadas.
MétodoEstudio epidemiológico transversal, periodo 2010-2015. Se captaron los posibles casos mediante los códigos 275.1 (CIE-9-MC), E83.0 (CIE-10) y 905 ORPHA en 15 RAER y el Registro de Pacientes de Enfermedades Raras del Instituto de Salud Carlos III. Los diagnósticos fueron validados revisando la documentación clínica. Se calcularon el valor predictivo positivo (VPP) de las fuentes de información, la prevalencia, la mortalidad y la distribución de las características sociodemográficas.
ResultadosEl Conjunto Mínimo Básico de Datos (CMBD) fue la fuente de información más utilizada por los RAER (VPP=39,4%), seguida del Registro de Medicamentos Huérfanos (RMH) (VPP=81,9%). La Historia Clínica de Atención Primaria (HCAP) obtuvo un VPP del 55,9%. Las combinaciones con mayor VPP fueron las del RMH con el CMBD (VPP=95,8%) y del RMH con la HCAP (VPP=92,9%). Se confirmaron 514 casos, el 57,2% eran hombres, cuya edad mediana de diagnóstico fue de 21,3 años. La prevalencia fue de 1,64/100.000 habitantes en 2015 y la mortalidad del 3,0%, siendo ambas superiores en los hombres.
ConclusiónSe recomienda la incorporación en los RAER del RMH y de la HCAP, ya que su combinación y la del RMH con el CMBD podrían utilizarse como criterio de validación automática para la enfermedad de Wilson. La prevalencia obtenida fue similar a la de otros países próximos a España.
To evaluate the sources of information used by the Regional Population-based Registries of Rare Diseases (RRD) for Wilson's Disease identification in Spain; to calculate its prevalence and mortality; and to describe the sociodemographic characteristics of those affected.
MethodCross-sectional epidemiological study, period 2010-2015. Possible cases were identified by codes 275.1 (ICD-9-CM), E83.0 (ICD-10) and 905 (ORPHAcode) in: 15 participating RRD and the Rare Disease Patients Registry of the Carlos III Health Institute. The diagnoses were confirmed through a clinical documentation review. The positive predictive value (PPV) of the sources of information used by RRD and their combinations were obtained. The prevalence, mortality and the distribution of sociodemographic characteristics were calculated.
ResultsThe Hospital Discharge Database (HDD) was the most used source by the RRD (PPV=39.4%), followed by the Orphan Drugs Registry (ODR) (PPV=81.9%). The Clinical History of Primary Care (PC) obtains PPV=55.9%. The combinations with highest PPV were the ODR with HDD (PPV=95.8%) and the ODR with PC (PPV=92.9%). 514 cases were confirmed, 57.2% men, with a median age of diagnosis of 21.3 years. The prevalence was 1.64/100,000 inhabitants in 2015 and mortality rate was 3.0%, being both higher in men.
ConclusionsIncorporation of ODR and PC into the RRD is recommended, as its combination and ODR with HDD could be used as an automatic validation criterion for Wilson's disease. The prevalence obtained was similar to that of countries close to Spain.
La enfermedad de Wilson (EW) es una enfermedad rara autosómica recesiva causada por mutaciones del gen ATP7B. Cursa con alteración del metabolismo del cobre, provocando su acumulación (en especial en el hígado, el sistema nervioso central y la córnea) y causando múltiples manifestaciones clínicas, principalmente hepáticas, neurológicas y psiquiátricas1,2. Los síntomas varían en gran medida entre las personas afectadas e incluso existen algunas asintomáticas. La dificultad diagnóstica de la EW reside en la heterogeneidad de las manifestaciones clínicas y la ausencia de una única prueba de confirmación3. Para su diagnóstico se desarrolló la escala Leipzig, un sistema de puntuación según los signos y síntomas que se presentan.
El diagnóstico precoz es vital para iniciar el tratamiento y con ello evitar el progreso de las lesiones. Existen diversas terapias farmacológicas2,4, que incluyen algunos medicamentos huérfanos: acetato de zinc (Wilzin®), trientina y tetratiomolibdato.
La EW, como el resto de las enfermedades raras, tiene gran trascendencia en salud pública por su impacto en la salud (cronicidad, discapacidad o mortalidad asociadas) y por los elevados costes sanitarios, sociales y emocionales que produce en cada persona afectada y su entorno5,6.
La prevalencia de la EW estimada por Orphanet es de 1-9 por 100.000 habitantes7, y los estudios de prevalencia realizados en diferentes países reflejan esta alta variabilidad8,9. A nivel nacional no se han realizado estudios epidemiológicos poblacionales sobre la EW.
En 2015 se publicó el Real Decreto 1091/2015, por el que se crea y regula el Registro Estatal de Enfermedades Raras (ReeR)10, que recoge información de los Registros Autonómicos de Enfermedades Raras (RAER)11. La EW se incluye entre las diez enfermedades raras recogidas inicialmente por el ReeR, para las que existen criterios de validación de casos por los RAER a través de fichas de validación12, pero aún no hay datos nacionales disponibles. Los RAER usan diferentes fuentes de información para captar posibles casos según su disponibilidad, y además utilizan diferentes sistemas de codificación13.
Los objetivos de este trabajo fueron determinar el valor predictivo positivo (VPP) de las fuentes de información utilizadas por los RAER para la captación de posibles casos y de las combinaciones de dichas fuentes; identificar la prevalencia y la mortalidad; y describir las características sociodemográficas de los casos confirmados de EW para el periodo 2010-2015.
MétodoSe realizó un estudio epidemiológico observacional transversal de base poblacional del periodo 2010-2015. Entre 2017 y 2019 participaron 15 RAER de diferentes comunidades autónomas: Aragón, Principado de Asturias, Illes Balears, Canarias, Cantabria, Castilla y León, Castilla-La Mancha, Comunitat Valenciana, Extremadura, Galicia, Comunidad de Madrid, Región de Murcia, Comunidad Foral de Navarra, País Vasco y La Rioja. También participó el Instituto de Investigación de Enfermedades Raras, que coordina y dirige el Registro de Pacientes de Enfermedades Raras (RPER) del Instituto de Salud Carlos III, de ámbito nacional. El Área de Investigación en Enfermedades Raras de la Fundación para el Fomento de la Investigación Sanitaria y Biomédica de la Comunitat Valenciana fue el centro coordinador.
Las fuentes de captación utilizadas por los RAER fueron el Conjunto Mínimo Básico de Datos (CMBD), el Registro de Mortalidad, el Registro de Medicamentos Huérfanos (RMH), el Registro de Anomalías Congénitas, los Registros Institucionales Sociales y Educativos, los registros específicos de enfermedades raras de las comunidades autónomas, la historia clínica de atención primaria (HCAP) y otras fuentes.
Como criterios de inclusión se consideraron posibles casos los captados hasta el año 2015 por los RAER, residentes en la autonomía del RAER declarante e identificados por literal de la palabra «Wilson» o los códigos 275.1 de la Clasificación Internacional de Enfermedades 9.ª revisión Modificación Clínica (CIE-9-MC) o E83.0 de la CIE-10.ª revisión (CIE-10) correspondientes a «Trastornos del metabolismo del cobre», o código ORPHA 905 de Orphanet correspondiente a «Enfermedad de Wilson», o sujetos tratados con medicamentos huérfanos indicados en la EW en el RMH.
Dada la inespecificidad de algunos de estos códigos, fue necesario validar el diagnóstico de EW en los posibles casos captados mediante la revisión de la documentación clínica. Así, se clasificaron como caso confirmado, no caso o caso probable (cuando en el momento de la validación no existía información suficiente para verificar o descartar el diagnóstico).
Los sujetos excluidos fueron residentes fuera de España o en situación irregular, los fallecidos antes del 1 de enero de 2010 y aquellos cuyo diagnóstico no pudo ser validado o certificado. Los residentes en Andalucía y Cataluña no se incluyeron por no participar los RAER de estas comunidades autónomas en el estudio.
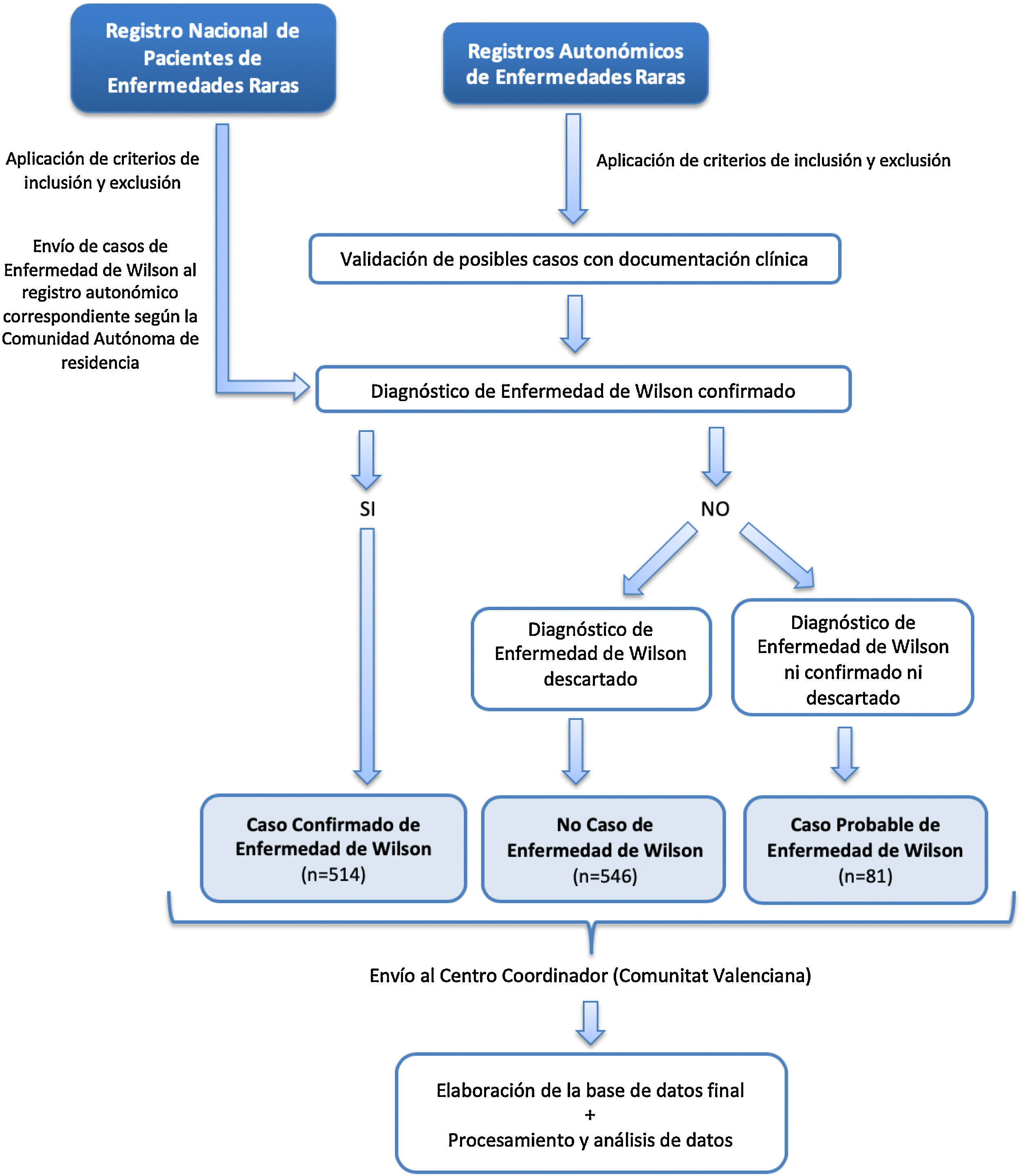
El esquema del proceso desde la captación de los posibles casos en los RAER hasta el análisis de los datos se recoge en la figura 1. Los casos del RPER se consideraron casos confirmados por tratarse de autodeclaración del paciente evaluada por profesionales mediante certificado médico acreditativo del diagnóstico. Se remitieron a los RAER, según el lugar de residencia, para depurar posibles duplicados.

Se calculó el VPP de las fuentes que utilizaron los RAER, por separado y en conjunto, dividiendo los casos confirmados entre los posibles casos captados. Se obtuvo el VPP de las combinaciones de las fuentes más utilizadas por los RAER (CMBD, RMH, Registro de Mortalidad y HCAP). Además, dado que pocos RAER disponían de HCAP, se analizó su potencial como fuente de captación identificando el porcentaje de casos confirmados captados exclusivamente por esta fuente.
Se calculó la prevalencia por 100.000 habitantes y su intervalo de confianza del 95% (IC95%) de los casos confirmados en total, por comunidades autónomas y por sexo, para cada año del periodo 2010-2015. La población de las comunidades autónomas participantes se obtuvo del Instituto Nacional de Estadística.
A partir de la información disponible en los RAER, procedente en algunos casos de los Registros de Mortalidad autonómicos, se analizó la mortalidad de la EW durante el periodo, su distribución por sexos, la edad mediana de defunción con su rango intercuartílico (RIC) y las principales causas de defunción, codificadas según la CIE-10.
Se realizó el análisis descriptivo de las características sociodemográficas de los casos confirmados: distribución por sexo, país de nacimiento y edad mediana en el momento del diagnóstico con su RIC. Para calcular la edad mediana se tomó la fecha de diagnóstico, y cuando esta se desconocía, la fecha de captación en el RAER.
Entre los no casos se identificaron las principales enfermedades confusoras. Se incluyó bajo el término «enfermedad confusora» cualquier trastorno asignado a los códigos correspondientes a EW que no se confirmó como EW tras la validación.
Galicia proporcionó datos agregados y, por tanto, solo se consideraron en el análisis para el cálculo del VPP del conjunto de fuentes de información y de la prevalencia. El País Vasco proporcionó el número de casos confirmados totales y el VPP de sus fuentes por separado, pero no el número de posibles casos captados ni las fuentes de captación de los casos confirmados, por lo que no fue posible analizar esta información individualmente. Extremadura no pudo aportar datos del año 2013.
El acceso a los datos se realizó por el personal propio de cada RAER, garantizando el cumplimiento de la normativa específica de cada registro y cumpliendo con la Ley Orgánica 15/1999, de 13 de diciembre, de Protección de Datos de Carácter Personal, vigente en el momento de realización del estudio.
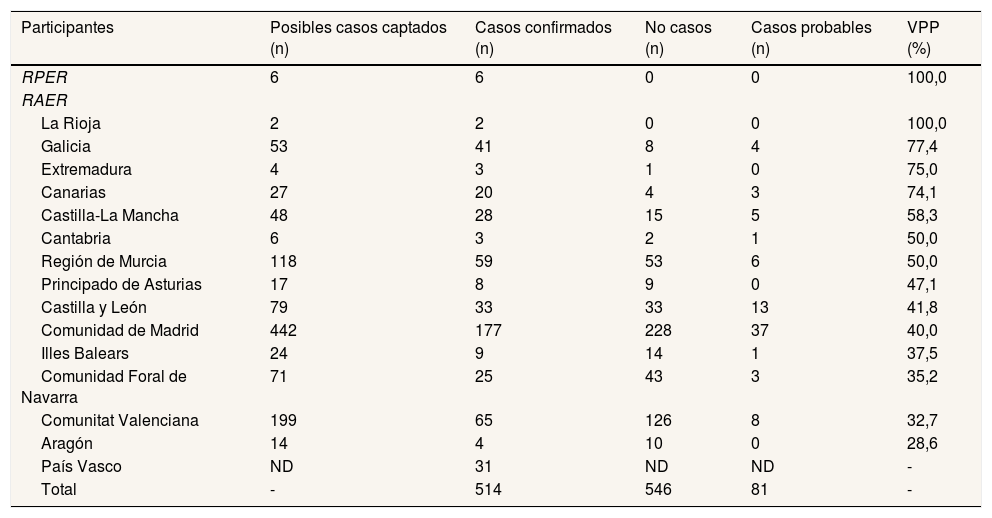
ResultadosSe identificaron 514 casos confirmados, 546 no casos y 81 casos probables (fig. 1). El VPP del conjunto de las fuentes de captación de cada RAER osciló entre el 28,6% en Aragón y el 100% en La Rioja (tabla 1).
Número de posibles casos captados, casos confirmados, no casos y casos probables, y valor predictivo positivo del conjunto de fuentes de los registros participantes en el estudio, para la enfermedad de Wilson, durante el periodo 2010-2015
| Participantes | Posibles casos captados (n) | Casos confirmados (n) | No casos (n) | Casos probables (n) | VPP (%) |
|---|---|---|---|---|---|
| RPER | 6 | 6 | 0 | 0 | 100,0 |
| RAER | |||||
| La Rioja | 2 | 2 | 0 | 0 | 100,0 |
| Galicia | 53 | 41 | 8 | 4 | 77,4 |
| Extremadura | 4 | 3 | 1 | 0 | 75,0 |
| Canarias | 27 | 20 | 4 | 3 | 74,1 |
| Castilla-La Mancha | 48 | 28 | 15 | 5 | 58,3 |
| Cantabria | 6 | 3 | 2 | 1 | 50,0 |
| Región de Murcia | 118 | 59 | 53 | 6 | 50,0 |
| Principado de Asturias | 17 | 8 | 9 | 0 | 47,1 |
| Castilla y León | 79 | 33 | 33 | 13 | 41,8 |
| Comunidad de Madrid | 442 | 177 | 228 | 37 | 40,0 |
| Illes Balears | 24 | 9 | 14 | 1 | 37,5 |
| Comunidad Foral de Navarra | 71 | 25 | 43 | 3 | 35,2 |
| Comunitat Valenciana | 199 | 65 | 126 | 8 | 32,7 |
| Aragón | 14 | 4 | 10 | 0 | 28,6 |
| País Vasco | ND | 31 | ND | ND | - |
| Total | - | 514 | 546 | 81 | - |
ND: datos no disponibles; RAER: Registros Autonómicos de Enfermedades Raras; RPER: Registro de Pacientes de Enfermedades Raras; VPP: valor predictivo positivo.
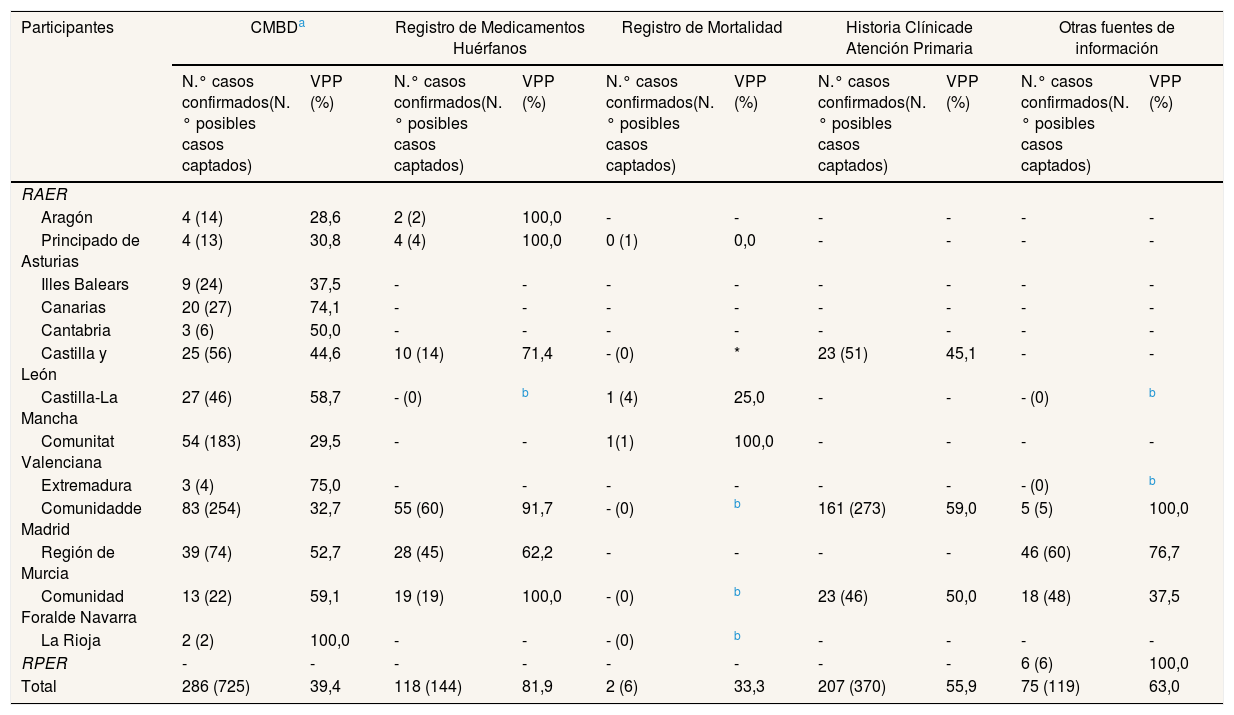
La tabla 2 muestra las fuentes de captación más empleadas por los RAER y su VPP. El CMBD (VPP=39,4%) fue la única fuente utilizada por todos los RAER; la segunda más utilizada fue el RMH (VPP=81,9%).
Número de posibles casos captados y casos confirmados, y valor predictivo positivo, para las fuentes de información más empleadas en la captación por los participantes en el estudio, para la enfermedad de Wilson, durante el periodo 2010-2015a
| Participantes | CMBDa | Registro de Medicamentos Huérfanos | Registro de Mortalidad | Historia Clínicade Atención Primaria | Otras fuentes de información | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| N.° casos confirmados(N.° posibles casos captados) | VPP (%) | N.° casos confirmados(N.° posibles casos captados) | VPP (%) | N.° casos confirmados(N.° posibles casos captados) | VPP (%) | N.° casos confirmados(N.° posibles casos captados) | VPP (%) | N.° casos confirmados(N.° posibles casos captados) | VPP (%) | |
| RAER | ||||||||||
| Aragón | 4 (14) | 28,6 | 2 (2) | 100,0 | - | - | - | - | - | - |
| Principado de Asturias | 4 (13) | 30,8 | 4 (4) | 100,0 | 0 (1) | 0,0 | - | - | - | - |
| Illes Balears | 9 (24) | 37,5 | - | - | - | - | - | - | - | - |
| Canarias | 20 (27) | 74,1 | - | - | - | - | - | - | - | - |
| Cantabria | 3 (6) | 50,0 | - | - | - | - | - | - | - | - |
| Castilla y León | 25 (56) | 44,6 | 10 (14) | 71,4 | - (0) | * | 23 (51) | 45,1 | - | - |
| Castilla-La Mancha | 27 (46) | 58,7 | - (0) | b | 1 (4) | 25,0 | - | - | - (0) | b |
| Comunitat Valenciana | 54 (183) | 29,5 | - | - | 1(1) | 100,0 | - | - | - | - |
| Extremadura | 3 (4) | 75,0 | - | - | - | - | - | - | - (0) | b |
| Comunidadde Madrid | 83 (254) | 32,7 | 55 (60) | 91,7 | - (0) | b | 161 (273) | 59,0 | 5 (5) | 100,0 |
| Región de Murcia | 39 (74) | 52,7 | 28 (45) | 62,2 | - | - | - | - | 46 (60) | 76,7 |
| Comunidad Foralde Navarra | 13 (22) | 59,1 | 19 (19) | 100,0 | - (0) | b | 23 (46) | 50,0 | 18 (48) | 37,5 |
| La Rioja | 2 (2) | 100,0 | - | - | - (0) | b | - | - | - | - |
| RPER | - | - | - | - | - | - | - | - | 6 (6) | 100,0 |
| Total | 286 (725) | 39,4 | 118 (144) | 81,9 | 2 (6) | 33,3 | 207 (370) | 55,9 | 75 (119) | 63,0 |
CMBD: Conjunto Mínimo Básico de Datos; RAER: Registros Autonómicos de Enfermedades Raras; RPER: Registro de Pacientes de Enfermedades Raras; VPP: valor predictivo positivo.
El VPP para las combinaciones de dos fuentes de captación fue del 95,8% para CMBD con RMH (casos confirmados=69), del 92,9% para RMH con HCAP (casos confirmados=65) y del 74,5% para CMBD con HCAP (casos confirmados=102). Las combinaciones del Registro de Mortalidad con CMBD, HCAP o RMH no aportaron casos confirmados adicionales.
El porcentaje de casos confirmados captados exclusivamente por la HCAP fue del 38,4% en la Comunidad de Madrid (casos confirmados=68), del 16,0% en la Comunidad Foral de Navarra (casos confirmados=4) y del 13,9% en Castilla y León (casos confirmados=5).
Los RAER que emplearon fuentes clasificadas como «otras» fueron Castilla-La Mancha (Registro de Enfermos Renales en Tratamiento Sustitutivo), Extremadura (Registros de Enfermos Renales en Tratamiento Sustitutivo y de Metabolopatías), Comunidad de Madrid (información proporcionada por el RPER), Región de Murcia (Medicamentos Extranjeros, Registro de Derivación de Pacientes a otras Regiones, Servicio de Valoración de Dependencia, Unidades de Hepatología y Genética) y Comunidad Foral de Navarra (Registro de Incapacidad Temporal y Servicio de Genética). El RPER también se consideró como «otras».
Las fuentes utilizadas por solo un RAER fueron Registros Específicos de Enfermedades Raras (Castilla-La Mancha, casos confirmados=28, VPP=58,3%), Registro de Anomalías Congénitas (Comunidad Foral de Navarra, casos confirmados=2, VPP=100%) y Registros Institucionales Sociales y Educativos (Región de Murcia, casos confirmados=1, VPP=100%).
En Galicia, las fuentes de captación de posibles casos fueron el CMBD, el RMH, la HCAP y otras (Laboratorio de Genómica y búsqueda de texto libre en documentación clínica). Las empleadas por el País Vasco fueron el CMBD (VPP=40%), la Historia Clínica de Consultas Especializadas (VPP=69%) y la notificación directa de casos por especialistas (VPP=100%).
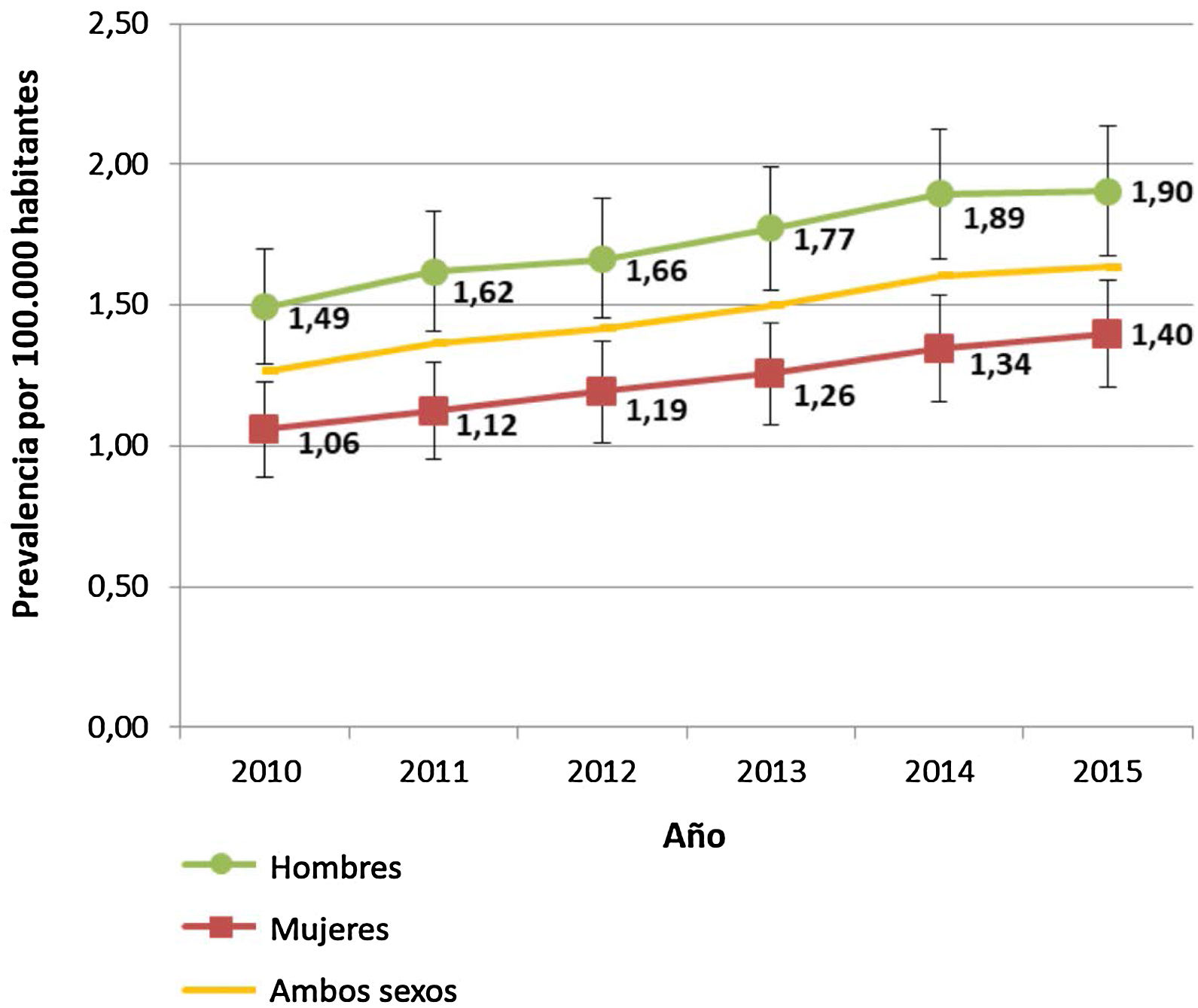
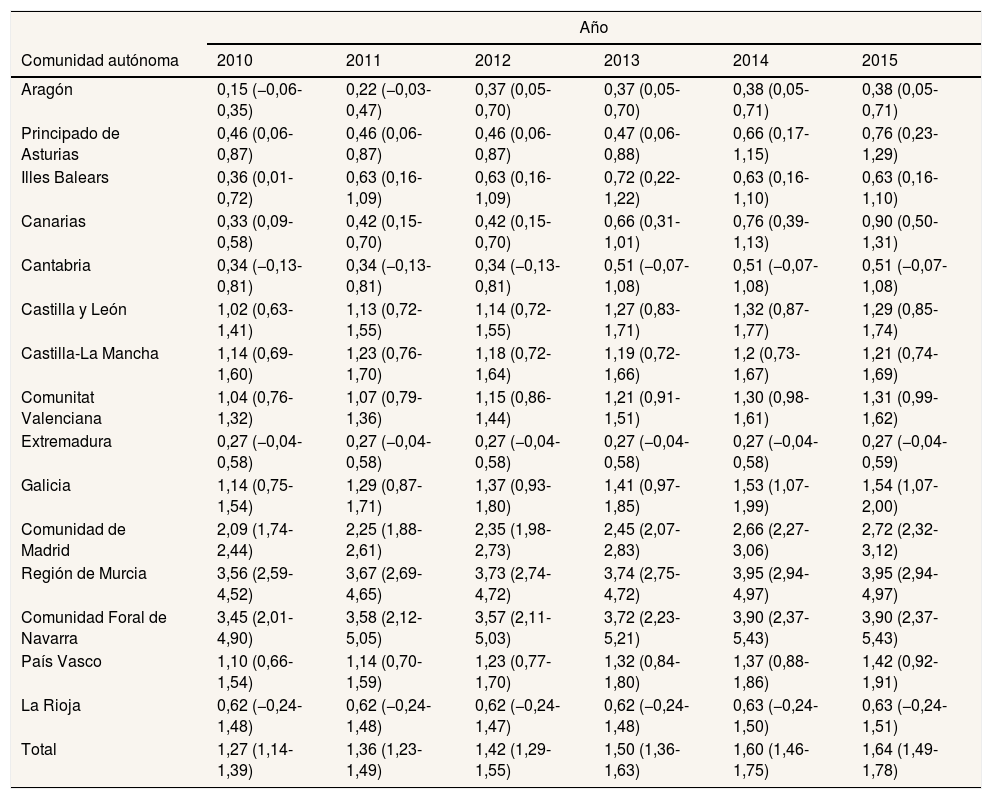
La prevalencia total de la EW para el conjunto de las comunidades autónomas participantes en 2015 fue de 1,64/100.000 habitantes (IC95%: 1,49-1,78). Por comunidades autónomas, esta prevalencia fue heterogénea: desde 0,27/100.000 en Extremadura hasta 3,95/100.000 en la Región de Murcia (tabla 3). Por sexos, la prevalencia fue mayor en los hombres durante todo el periodo estudiado, siendo la diferencia estadísticamente significativa (fig. 2).
Prevalencia anual por 100.000 habitantes e intervalo de confianza del 95% según la comunidad autónoma de residencia y prevalencia anual total, para la enfermedad de Wilson, durante el periodo 2010-2015
| Año | ||||||
|---|---|---|---|---|---|---|
| Comunidad autónoma | 2010 | 2011 | 2012 | 2013 | 2014 | 2015 |
| Aragón | 0,15 (−0,06-0,35) | 0,22 (−0,03-0,47) | 0,37 (0,05-0,70) | 0,37 (0,05-0,70) | 0,38 (0,05-0,71) | 0,38 (0,05-0,71) |
| Principado de Asturias | 0,46 (0,06-0,87) | 0,46 (0,06-0,87) | 0,46 (0,06-0,87) | 0,47 (0,06-0,88) | 0,66 (0,17-1,15) | 0,76 (0,23-1,29) |
| Illes Balears | 0,36 (0,01-0,72) | 0,63 (0,16-1,09) | 0,63 (0,16-1,09) | 0,72 (0,22-1,22) | 0,63 (0,16-1,10) | 0,63 (0,16-1,10) |
| Canarias | 0,33 (0,09-0,58) | 0,42 (0,15-0,70) | 0,42 (0,15-0,70) | 0,66 (0,31-1,01) | 0,76 (0,39-1,13) | 0,90 (0,50-1,31) |
| Cantabria | 0,34 (−0,13-0,81) | 0,34 (−0,13-0,81) | 0,34 (−0,13-0,81) | 0,51 (−0,07-1,08) | 0,51 (−0,07-1,08) | 0,51 (−0,07-1,08) |
| Castilla y León | 1,02 (0,63-1,41) | 1,13 (0,72-1,55) | 1,14 (0,72-1,55) | 1,27 (0,83-1,71) | 1,32 (0,87-1,77) | 1,29 (0,85-1,74) |
| Castilla-La Mancha | 1,14 (0,69-1,60) | 1,23 (0,76-1,70) | 1,18 (0,72-1,64) | 1,19 (0,72-1,66) | 1,2 (0,73-1,67) | 1,21 (0,74-1,69) |
| Comunitat Valenciana | 1,04 (0,76-1,32) | 1,07 (0,79-1,36) | 1,15 (0,86-1,44) | 1,21 (0,91-1,51) | 1,30 (0,98-1,61) | 1,31 (0,99-1,62) |
| Extremadura | 0,27 (−0,04-0,58) | 0,27 (−0,04-0,58) | 0,27 (−0,04-0,58) | 0,27 (−0,04-0,58) | 0,27 (−0,04-0,58) | 0,27 (−0,04-0,59) |
| Galicia | 1,14 (0,75-1,54) | 1,29 (0,87-1,71) | 1,37 (0,93-1,80) | 1,41 (0,97-1,85) | 1,53 (1,07-1,99) | 1,54 (1,07-2,00) |
| Comunidad de Madrid | 2,09 (1,74-2,44) | 2,25 (1,88-2,61) | 2,35 (1,98-2,73) | 2,45 (2,07-2,83) | 2,66 (2,27-3,06) | 2,72 (2,32-3,12) |
| Región de Murcia | 3,56 (2,59-4,52) | 3,67 (2,69-4,65) | 3,73 (2,74-4,72) | 3,74 (2,75-4,72) | 3,95 (2,94-4,97) | 3,95 (2,94-4,97) |
| Comunidad Foral de Navarra | 3,45 (2,01-4,90) | 3,58 (2,12-5,05) | 3,57 (2,11-5,03) | 3,72 (2,23-5,21) | 3,90 (2,37-5,43) | 3,90 (2,37-5,43) |
| País Vasco | 1,10 (0,66-1,54) | 1,14 (0,70-1,59) | 1,23 (0,77-1,70) | 1,32 (0,84-1,80) | 1,37 (0,88-1,86) | 1,42 (0,92-1,91) |
| La Rioja | 0,62 (−0,24-1,48) | 0,62 (−0,24-1,48) | 0,62 (−0,24-1,47) | 0,62 (−0,24-1,48) | 0,63 (−0,24-1,50) | 0,63 (−0,24-1,51) |
| Total | 1,27 (1,14-1,39) | 1,36 (1,23-1,49) | 1,42 (1,29-1,55) | 1,50 (1,36-1,63) | 1,60 (1,46-1,75) | 1,64 (1,49-1,78) |

La mortalidad fue del 3,0%, y el 71,4% de los fallecidos fueron hombres. La edad mediana de defunción fue de 47,5 años (RIC: 45,5-62,7). Entre las causas de defunción conocidas destacaron la EW (14,3%) y la neoplasia maligna de hígado y de vías biliares intrahepáticas (14,3%), la cirrosis hepática (7,1%) y la neoplasia maligna de bronquio y pulmón (7,1%). En el 57,2% la causa de la defunción fue desconocida.
Respecto a las características sociodemográficas, los casos confirmados fueron el 57,2% hombres y el 42,8% mujeres. La mayoría (86,0%) habían nacido en España, seguidos de Rumanía (1,5%), China (1,3%) y Ecuador (1,3%). El 3,4% nacieron en otros países y en el 6,5% se desconocía esta información. La edad mediana en el momento del diagnóstico fue de 21,3 años (RIC: 12,0-36,1). Las enfermedades confusoras identificadas en los no casos y su distribución se recogen en la tabla 4.
Distribución de los no casos según la enfermedad confusoraa identificada tras la validación, para la enfermedad de Wilson, durante el periodo 2010-2015
| Enfermedad confusora | No casos | Porcentaje |
|---|---|---|
| Valores bajos de cobre en sangre | 65 | 11,9 |
| Alteraciones hepáticas (esteatosis, enfermedad alcohólica del hígado, cirrosis, etc.) | 64 | 11,7 |
| Estudio de portadores genéticos | 57 | 10,4 |
| Valores anormales de enzimas en suero (transaminasas o ceruloplasmina) | 29 | 5,3 |
| Carencia nutricional de cobre | 33 | 6,0 |
| Síndrome de Gilbert | 13 | 2,4 |
| Neoplasias | 13 | 2,4 |
| Trastornos del metabolismo del hierro | 11 | 2,0 |
| Trastornos de la conducta alimentaria | 8 | 1,5 |
| Cirugía bariátrica | 7 | 1,3 |
| Trastornos del metabolismo del cobre diferentes de la enfermedad de Wilson | 5 | 0,9 |
| Malabsorción intestinal | 2 | 0,4 |
| Otras enfermedades confusoras | 66 | 12,1 |
| Enfermedad confusora no identificada | 165 | 30,2 |
| No disponibleb | 8 | 1,5 |
| Total | 546 | 100,0 |
Este es el primer estudio epidemiológico de base poblacional en España sobre la EW con participación de casi todas las comunidades autónomas y el RPER, representando el 65,9% de la población en 2015.
Entre las fuentes de captación, el CMBD fue la única utilizada por todos los RAER. El RMH obtuvo el mayor VPP. La EW dispone de medicamentos huérfanos para su tratamiento2,4, lo que explicaría este elevado VPP, aunque algunos de estos fármacos tienen otras indicaciones además de la EW y por ello el VPP no alcanza el 100%.
La combinación del CMBD con el RMH obtuvo un elevado VPP, haciendo posible la validación directa (sin revisión de la documentación clínica) de todos los posibles casos captados por ambas fuentes. La ficha de validación de la EW del ReeR únicamente considera como criterio de validación directa la combinación del CMBD con el medicamento huérfano Wilzin®, pero el resultado obtenido en este estudio permitiría ampliar este criterio al resto de los medicamentos huérfanos con indicación para la EW. El RMH junto con la HCAP también alcanzó un VPP elevado, por lo que podría añadirse como criterio de validación en la ficha, aunque su uso no está muy extendido. Finalmente, la combinación del CMBD con la HCAP no obtuvo un valor adecuado como para considerarla criterio de validación, pero sería interesante evaluar en un futuro las combinaciones de más de dos fuentes.
Respecto al uso de la HCAP, los resultados mostraron que el porcentaje de casos confirmados captados únicamente por esta fuente fue considerable. Además, el CMBD recoge diagnósticos asociados a altas hospitalarias, y por tanto los casos que no requirieron ingreso no fueron captados por los RAER que no incluyeron la HCAP. En cambio, los RAER que sí la utilizaron pudieron captarlos, lo que explica el notable porcentaje de casos confirmados aportados exclusivamente por la HCAP. Por otra parte, el RMH, pese a su elevado VPP, solo está disponible en ocho de los RAER participantes. Por tanto, generalizar el empleo del RMH y la HCAP como fuentes de captación en los RAER podría suponer un incremento de los casos captados. Además, en el ámbito nacional, está previsto que a corto plazo los Centros, Servicios y Unidades de Referencia (CSUR) sean una fuente de captación más en los RAER, lo que permitirá mejorar la captación de casos con menor expresividad clínica.
El RPER podría incluirse como fuente de casos validada en el ReeR, ya que aporta casos confirmados mediante certificado médico del diagnóstico.
La prevalencia total obtenida se encuentra dentro del rango estimado por Orphanet7 y fue similar a la descrita en Francia en 2013 (1,5/100.000 habitantes)8, donde utilizaron una metodología similar, pero sin validación con documentación clínica. Tanto el estudio francés8 como un metaanálisis9 recogieron resultados muy heterogéneos de prevalencia en diversos países, desde valores antiguos, como los de Alemania del Este en 1974 (0,46/100.000)14, Escocia en 1989 (0,4/100.000)15, Cerdeña en 1983 (2,9/100.000)16, Israel en 1958-1985 (0,25/100.000)17 y Albania en 1991 (0,68/100.000)18, hasta más recientes, como los de Irlanda en 2011 (0,9/100.000)19, Taiwán en 2005 (1,60/100.000)20 y en 2000-2011 (1,81/100.000)21, Dinamarca en 1990-2008 (2,02/100.000)22 y la región china de Anhui en 2008-2011 (5,87/100.000)23.
Esta gran variabilidad podría deberse a los diferentes métodos utilizados y a la mejora del diagnóstico de la EW, como sugiere el estudio francés8, justificando la menor prevalencia en los estudios más antiguos. En España, la prevalencia en las diferentes comunidades autónomas también resultó heterogénea, hecho que podría explicarse por las distintas fuentes de captación utilizadas por los RAER; por ejemplo, el País Vasco dispone de notificación directa por parte de médicos especialistas y de captación en las historias clínicas de consultas especializadas, y otras comunidades autónomas disponen de búsqueda de texto libre en documentación clínica, como Navarra y Galicia. Muchos valores de prevalencia bajos corresponden a comunidades autónomas cuyos RAER solo utilizaron el CMBD como fuente de captación, mientras que las dos con mayor prevalencia (Murcia y Navarra) son las únicas con información específica procedente de laboratorios de genética. Esta variabilidad entre comunidades autónomas no debería atribuirse a factores de atracción sanitaria hacia centros de referencia, ya que se han incluido los residentes en la comunidad autónoma declarante.
Asimismo, se observó un ascenso significativo en la prevalencia total, que podría explicarse por el mayor conocimiento de la EW por parte del personal sanitario y, en consecuencia, el mejor diagnóstico y adecuado tratamiento de las personas afectadas. Además, en zonas aisladas la prevalencia podría ser más alta debido a la consanguinidad y la mayor frecuencia de determinadas mutaciones24,25.
Por sexos, se encontró una prevalencia significativamente mayor en los hombres durante el periodo estudiado, en concordancia con estudios realizados en Francia8, Taiwán26 y Polonia27. Aunque en Alemania28 y Austria29 se han hallado resultados opuestos, sus poblaciones de estudio no fueron identificadas mediante registros poblacionales.
En Francia8, la mortalidad fue del 3,1%, similar a la de este estudio, pero en Alemania28 fue del 1,8%, en Austria29 del 7,4% y en Polonia30 del 12,0%. Por tanto, se observa una disparidad entre países que también podría deberse a las diferencias metodológicas, sobre todo en la selección de la población de estudio.
En España, entre las causas conocidas de defunción predominaron la propia EW y problemas hepáticos relacionados probablemente con la EW, ya que las principales causas de fallecimiento de estos enfermos suelen ser complicaciones hepáticas2. No obstante, se deberán confirmar estos resultados, pues la causa de la muerte era desconocida en más de la mitad de los casos fallecidos.
La mayoría de los fallecidos fueron hombres y, teniendo en cuenta que la prevalencia en ellos también fue mayor, sería necesario abordar futuras investigaciones centradas en las diferencias entre sexos, ya que otros estudios han encontrado diferencias relacionadas con la presentación hepática o neurológica de la enfermedad27 y en la ocurrencia de fallo hepático fulminante28,29.
La mediana de edad en el momento del diagnóstico fue de 21,3 años. En Alemania28 la edad media fue de 20,4 años, en Polonia27 de 28,0 años y en Austria29 de 21,2 años (presentación hepática) y 28,8 años (presentación neurológica). Pese a utilizar diferentes poblaciones de estudio, parece que en la edad de diagnóstico existe menos variabilidad y se sitúa siempre alrededor de la tercera década de la vida.
Entre las enfermedades confusoras identificadas en los no casos se encontraron trastornos cuyos signos y síntomas podrían atribuirse a la EW, pudiendo ser esta la sospecha diagnóstica en algún momento. No obstante, estos trastornos disponen de códigos específicos en la CIE-9-MC y la CIE-10, y por tanto una correcta codificación aumentaría el VPP de las fuentes de captación (en concreto del CMBD y la HCAP), reduciendo el volumen de validación. Además, el 10,4% de no casos fueron estudios genéticos de la EW en parejas o familiares de personas afectadas, que son codificados como EW en la fuente original, pero no se pueden confirmar ni descartar como EW hasta su validación. Aunque su captación conllevó la necesidad de validación, resulta interesante identificar cuántas personas se sometieron a estos estudios. Solo el 0,9% de los no casos fueron finalmente diagnosticados de algún trastorno del metabolismo del cobre distinto de la EW, y por tanto estaban correctamente codificados.
Respecto a las limitaciones, el RAER de Extremadura no aportó datos de 2013, aunque dada la baja prevalencia identificada en esta comunidad autónoma no se consideró que pudiera ser relevante para la prevalencia total. Por otra parte, la inespecificidad de los códigos CIE-9-MC y CIE-10 utilizados para captar posibles casos obligó a la validación del diagnóstico y podría ser la causa, en parte, del elevado número de no casos identificados. A partir de 2016, el CMBD empezó a codificar según la CIE-10-ES, que incorpora un código específico para la EW (E83.01), lo que previsiblemente mejorará el VPP de esta fuente (aunque la validación seguirá siendo necesaria al incluir bajo este código las sospechas diagnósticas). Finalmente, la heterogeneidad de las fuentes de captación y de sus correspondientes sistemas de codificación en los diferentes RAER podría explicar, en parte, la variabilidad del VPP obtenido para el conjunto de fuentes de cada RAER. Por ello, se realizó el análisis individualizado de fuentes.
Las fortalezas de este estudio son el hecho de tratarse de un estudio multicéntrico, la utilización de registros poblacionales y la verificación del diagnóstico mediante documentación clínica, excluyendo errores de codificación o sujetos con otros trastornos recogidos bajo los mismos códigos.
En conclusión, este trabajo confirma que los RAER son herramientas de gran utilidad en los estudios sobre la EW. No obstante, debería extenderse el uso de las fuentes de información con mejor VPP (principalmente el RMH y la HCAP) en todos los RAER, para identificar el mayor número posible de personas afectadas por la EW en nuestro país. Se han calculado la prevalencia y la mortalidad, se ofrece una visión diferenciada por comunidades autónomas y se identifican aquellas con mayor presencia de EW. Se ha evidenciado la tendencia ascendente en la prevalencia total debida probablemente al mejor diagnóstico y el mayor acceso al tratamiento, y se han puesto de manifiesto la prevalencia más alta y la mayor mortalidad de la EW en los hombres. Con esta información se pretende facilitar el desarrollo de políticas sanitarias de prevención y promover la investigación traslacional de la EW, para mejorar la calidad asistencial y la atención sociosanitaria de las personas afectadas.
La enfermedad de Wilson es una enfermedad rara de prevalencia muy variable según los países y las regiones donde se ha estudiado. En España se creó en 2015 el Registro Estatal de Enfermedades Raras, pero aún no se dispone de información publicada sobre la enfermedad ni tampoco se han evaluado las fuentes de información empleadas para su detección.
¿Qué añade el estudio realizado a la literatura?Este estudio pone de relevancia la importancia de las fuentes de información empleadas en investigación epidemiológica para aumentar el conocimiento sobre las enfermedades raras. Muestra los primeros datos epidemiológicos de la enfermedad de Wilson en España y con ello permite mejorar la calidad de los registros autonómicos de enfermedades raras, la atención a las personas afectadas y el diseño de estrategias de prevención.
Andreu Segura.
Declaración de transparenciaLa autora principal (garante responsable del manuscrito) afirma que este manuscrito es un reporte honesto, preciso y transparente del estudio que se remite a Gaceta Sanitaria, que no se han omitido aspectos importantes del estudio, y que las discrepancias del estudio según lo previsto (y, si son relevantes, registradas) se han explicado.
Contribuciones de autoríaC. Cavero-Carbonell concibió y diseñó el estudio. S. Moreno-Marro, L. Barrachina-Bonet, V. Alonso-Ferreira, M. García-López, J. Palomar-Rodríguez, E. Vicente y A. Clara Zoni realizaron el trabajo de recogida de datos. S. Moreno-Marro y L. Barrachina-Bonet se encargaron de la revisión, la integración y la adecuación de los datos. V. Alonso-Ferreira, M. García-López, J. Palomar-Rodríguez, E. Vicente y A. Clara Zoni resolvieron todas las dudas y aclaraciones pertinentes para poder garantizar la correcta integración de los datos. S. Guardiola-Vilarroig y O. Zurriaga facilitaron los materiales, los permisos y las autorizaciones necesarios para la correcta realización del trabajo. S. Moreno-Marro y L. Barrachina-Bonet trabajaron de forma activa en el análisis de los datos obtenidos y en la elaboración de los primeros resultados. L. Páramo-Rodríguez, S. Guardiola-Vilarroig y O. Zurriaga interpretaron los resultados y aportaron ideas para la redacción del manuscrito. S. Moreno-Marro redactó el primer borrador del manuscrito. C. Cavero-Carbonell, L. Barrachina-Bonet, V. Alonso-Ferreira, M. García-López, J. Palomar-Rodríguez, E. Vicente y A. Clara Zoni realizaron una revisión crítica con importantes contribuciones intelectuales. Todas las personas firmantes han hecho importantes aportaciones en los resultados y en su análisis, han contribuido con su bagaje intelectual a la redacción final del manuscrito y han aprobado la versión final. S. Moreno-Marro es la responsable del artículo.
AgradecimientosAgradecemos la colaboración del resto de las personas integrantes del Grupo de Trabajo Wilson-RAER (Registros poblacionales Autonómicos de Enfermedades Raras): Federico Arribas Monzón (Aragón), Mario Margolles Martins (Principado de Asturias), Mercedes Cáffaro Rovira (Illes Balears), Patricia Carrillo Ojeda (Canarias), Miguel García Ribes y Andrés Alvarado García (Cantabria), Marta Sedano Valdivieso y Pilar Peces Jimeno (Castilla-La Mancha), Rufino Álamo Sanz (Castilla y León), Julián Mauro Ramos Aceitero (Extremadura), Consuelo Benito Torres (Galicia), Antonia Sánchez Escámez (Región de Murcia), Guillermo Ezpeleta (Navarra), Luis María Oregi Lizarralde (País Vasco) y Enrique Ramalle Gomara (La Rioja). Su participación en el grupo de trabajo ha hecho posible este estudio. También agradecemos la colaboración con fondos de la Obra Social “laCaixa”.
FinanciaciónEste proyecto ha sido posible gracias a los fondos recibidos por la Fundació Per Amor a l’Art (Convenio CPRESC00043).
Conflictos de interesesNinguno.











